近日,我校生命科學學院計算與整合生物學團隊在國際基因組學領域權威學術期刊《Genome Biology》(IF=12.3,生物學一區Top)上在線發表了題為“Systematic evaluation with practical guidelines for single-cell and spatially resolved transcriptomics data simulation under multiple scenarios”的研究論文,對團隊在單細胞RNA測序和空間轉錄組學方法學領域取得的重要進展進行了介紹。該研究全面評估了單細胞/空間轉錄組數據模擬算法在多個流行應用場景下的表現,并為該領域的科研人員提供了詳盡的方法選擇使用指南。

重慶師范大學為論文第一完成單位和唯一通訊單位,生命科學學院2022級碩士研究生朵泓睿為第一作者,李勃副教授和郝友進教授為通訊作者,重慶郵電大學、陸軍軍醫大學、浙江大學、重慶大學和重慶醫科大學等單位的研究人員共同參與了此項工作。
近年來,隨著單細胞RNA測序(scRNA-seq)和空間轉錄組學(SRT)的迅速興起,兩者通過精確揭示單細胞水平基因表達的異質性及其在組織中的空間分布特征,顯著加速了現代生命科學的研究進程,推動了研究范式的轉變和創新。在scRNA-seq和SRT算法設計和數據分析工具基準測試中,模擬數據至關重要。盡管目前已有大量用于模擬scRNA-seq和SRT數據的方法,但面對具體的生物信息學分析任務時,如何選擇最適合的方法仍然是一個棘手的問題。
在該項研究中,團隊成員利用152組真實參考數據集對49種單細胞/空間轉錄組數據模擬方法在準確性、功能性、可拓展性和適用性方面開展了大規模的系統性評估。研究結果表明,建立在最優選擇模型(e.g., SRTsim 和 scDesign2)和 GAMLSS(e.g., scDesign3)基礎上的方法具有最佳的準確性表現,但需要以消耗更多時間和內存來建立基因表達數據模型為代價。此外,ZINB- WaVE、SPARSim、Splat、SCRIP-paths、muscat 和 SCRIP-GP-trendedBCV 也表現出較強的數據模擬能力。專用于scRNA-seq數據模擬的方法Splat、SPARSim、SCRIP、SplatPop、dropim 和 ZINB-WaVE 在模擬 SRT 數據方面具有強大的兼容性。在功能性方面,Lun在模擬細胞類群和差異表達基因的應用場景下的效果最佳,而 SPARSim和scDesign3-tree 則分別在細胞批次和軌跡模擬方面表現優于其他方法。由于沒有一種方法在所有評價標準上都表現出色,因此用戶應考慮在準確性和功能性、準確性和可擴展性之間做出權衡。在適用性方面,半數以上的方法在運行過程中會產生錯誤,而且錯誤比例在不同方法間具有較大差異。“基因表達值擬合失敗”和 “出現缺失(無窮)值 ”是運行失敗的兩個主要原因。
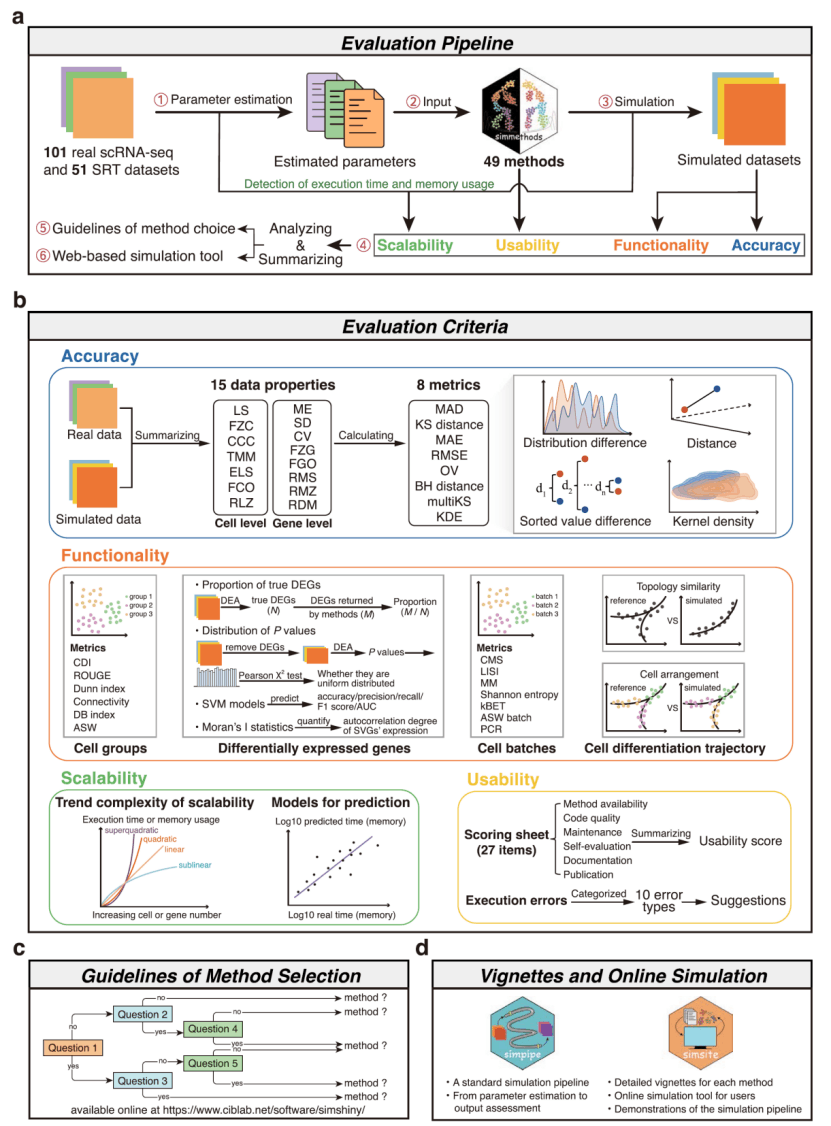
根據評估結果,團隊成員還建立了方法選擇的實用指南、標準數據模擬流程Simpipe和交互工具 Simsite(https://www.ciblab.net/software/Simsite/),以供用戶選擇合適的方法并執行模擬任務。這項研究將為單細胞組學領域的研究者提供指導和建議,幫助他們選擇合適的數據模擬方法。同時,它也將激勵算法開發人員提出更具擴展性和效率的方法,從而有助于研究者深刻理解和應用基因表達數據的特征。
據悉,《Genome Biology》是國際基因組生物學領域頂級學術期刊,主要發表從基因組和后基因組的角度研究生命科學的最新研究成果,創刊以來刊登過多項重大的生命科學研究進展,年刊載文章數為200-300篇。該期刊最新影響因子12.3,其5年影響因子為17.4,目前為中科院一區TOP期刊。
原文鏈接






